The central dogma of biology states that DNA is transcribed into RNA which in turn serves as the template for the translation to protein. According to the canonical view, a ribosome reads units of three RNA nucleotides (“codons”) and translates each of these into an amino acid. While theoretically, each RNA sequence can be read in three different frames and therefore encode three different proteins, usually only one of these reading frames is utilized. In the last decades, new findings revealed additional possibilities how the existing genetic information is translated into protein products. A recent study characterizing the coding capacity of SARS-CoV-2 found experimental evidence for the existence of 23 unannotated viral ORFs translated into protein, in addition to the twelve previously known ones (Finkel et al., 2021). This strongly suggests that only a small part of the genes encoded by the virus has been identified, limiting our ability to understand the devastating effect of viral infection.
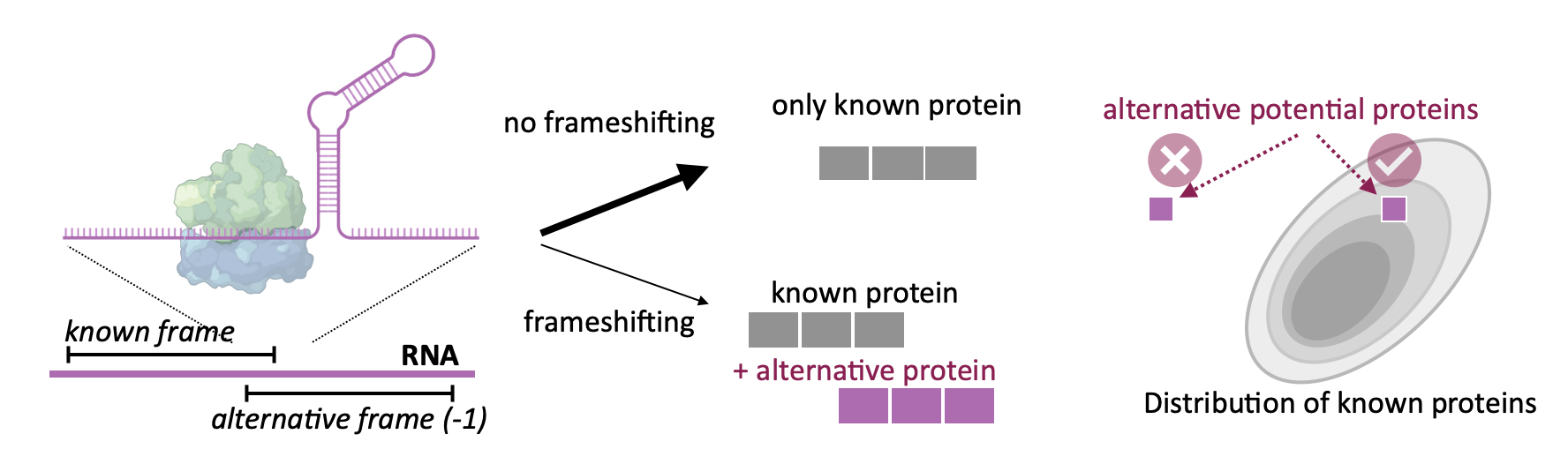
How can viruses and human cells generate alternative, still undiscovered proteins? One non-canonical mechanism of gene expression first discovered in viruses is programmed ribosomal frameshifting: a controlled slippage of the translating ribosome to an alternative reading frame at a defined position, generating two proteins from the same mRNA. This mechanism is crucial for viral replication, presenting it as a promising antiviral drug target. The extent, to which this or other mechanisms are used in different genomes is still unknown.
Here, we set out to identify alternative proteins resulting from non-canonical translation, which resemble functional proteins and therefore could perform important cellular functions. In this project, we developed a computational strategy – in combination with experiments testing the in silico predictions – to determine which alternative proteins could be encoded by a given sequence by shifting of the ribosome to a different reading frame. To this end, we used deep neural networks to predict if an amino acid sequence is a valid protein. Based on the vast databases of known proteins available to date we trained Variational Auto Encoders to learn the representation of possible proteins. We then used the trained model to obtain probabilities that non-canonical frame-shifted sequences are drawn from the same distribution of known functional proteins.
While this is an extraordinarily challenging task, we were able to obtain encouraging preliminary results already over the course of the seed funding period. In future our classifier of valid vs. non-valid proteins can be readily applied to a wide range of biological questions and can help exploring the many unknowns of the human genome.