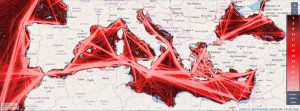
The data revolution and the increase in information and communication technologies elevate public interest and make everyone in academia, industry, non-profit organizations and the public sector a developer and/or user of data science (DS) methods and tools.
The University of Haifa (UoH) has an ideal combination of research disciplines in both core and applications of DS.
The proposed DS research center of the UoH (DS-RC@UoH) will integrate the core areas, such as machine learning, deep learning, big data, distributed algorithms, statistical inference and methodology, big data management, etc., and applications that use these methodologies, such as computer vision, natural language processing, computational biology, informatics, astronomy, etc.
The primary objective of the university-wide overarching center is to:
- Develop a mechanism to advance methodological DS research in an integrative/harmonized manner across all the faculties, schools, departments and DS-related centers;
- Increase the usage of DS methods and tools across all departments and disciplines;
- Boost collaboration with industry and the public sector in order to address real-life problems to better society.
- Enhance the transformation of the University of Haifa to the digital Era, digital sciences, digital humanities, and digital social sciences. We believe that the world and the academia as part of it are going through major transformations, and in the near future all researchers will need to comprehend DS technologies to stay in the leading edge of research.
To address its mission, the center will increase opportunities for collaboration among core DS scientists, DS scientists who are developing methods for specific application domains, and researchers in non-DS domains. The latter are users of DS tools who can define new needs and challenges that may spark innovative DS methodologies and step up the DS research in their domains (e.g. humanities, social sciences, etc.). Second, DS members who are established in working with and attaining data from the industry and the public sectors will develop strategies and activities to facilitate collaboration of additional researchers with these sectors.